
This two-day workshop will address some of the key product development and regulatory assessment considerations for generic glucagon-like peptide-1 (GLP-1) agonist products. The goal of this workshop is to provide clarity and foster discussions on evidence-based approaches to establish the acceptable critical quality attributes of recombinant and synthetic peptide products, address manufacturing quality considerations, and tackle the unique challenges associated with drug-device combination products, where applicable.
Key areas of focus include:
- Regulatory and Scientific Foundations: Current FDA scientific thinking on generic GLP-1 product development, Product-Specific Guidance (PSG) development, and in vivo BE studies for GLP-1 oral products
- Active Pharmaceutical Ingredient (API) Characterization and Manufacturing: Comprehensive approaches to demonstrating API sameness, analytical characterization strategies, impurity profiling, and sameness assessment for peptide products
- Recombinant Peptide Production: Best practices for controlling host cell impurities, regulatory expectations, and risk assessment strategies
- Oral GLP-1 Formulation Development: Formulation and delivery innovations, bioequivalence study design for highly variable products, absorption enhancement technologies, and drug product quality expectations
- Device User Interface and Human Factors: Comparative analyses (CA) of device user interfaces, Comparative Use Human Factors (CUHF) study design and case examples, alternative approaches, and device platform strategies
- Device Quality and Integration: Quality considerations for generic injectors, drug-device interface and compatibility, manufacturing quality control, and submission coordination strategies
Join us for an immersive two-day experience featuring presentations from leading experts in regulatory agencies, pharmaceutical industry, and academia. All attendees will benefit from insights shared by a diverse group of speakers, including FDA officials, industry leaders, and renowned academics. This workshop offers a unique opportunity to:
- Understand the latest regulatory thinking on generic GLP-1 product development
- Learn about cutting-edge analytical and formulation approaches for peptide products
- Gain insights into device design, human factors, and combination product development
- Network with peers and experts in the field
- Explore real-world case examples and best practices
- Participate in collaborative discussions on emerging challenges
Join us to be part of the conversation shaping the future of generic GLP-1 product development.
Choose your experience:
Virtual Attendees will have free access to all sessions of the workshop except the small group (in-person only) working sessions. Virtual attendees will be able to:
- Attend all presentations and panel discussions
- Participate in all Q&A panel discussions by submitting questions online in real time to the speakers and panelists
- Enjoy free access to workshop recordings of presentations and panel discussion (not including the small group working sessions)
In-person Attendees will enjoy all of the benefits of virtual attendance as well as interactive experiences featuring:
- Small group working sessions at the end of each day will facilitate collaborative problem-solving and discussion of complex technical and regulatory issues impacting GLP-1 generics. During these working sessions, attendees will help identify actionable next steps to address challenges with generic product development and regulatory assessment.
- Collaborative interactions with experts from industry, consulting groups, academia and FDA for clarifying regulatory expectations and exploring insights into regulatory standards
- Networking opportunities designed to foster collaboration and contribute to moving the field forward.
Workshop Topics
- Regulatory Pathways and Clinical Context
- API Characterization and Peptide Manufacturing
- Oral Formulation Development and Bioequivalence
- Device Design and Human Factors
- Drug-Device Integration and Quality
Audience
This workshop is designed for professionals involved in the development, manufacturing, and regulation of generic GLP-1 products. The target audience includes pharmaceutical scientists (formulation and product development scientists, analytical chemists, quality assurance and control experts), regulatory affairs professionals, device engineers, human factors specialists, clinical development professionals, and manufacturing experts. Additionally, the workshop will be valuable for academic researchers in pharmaceutical sciences, representatives from contract research organizations (CROs) and contract manufacturing organizations (CMOs), regulatory agency staff, and healthcare professionals interested in generic complex drug products. The content is tailored to accommodate participants with varying levels of expertise in peptide products and drug-device combination products, from those seeking to understand the fundamentals to advanced practitioners looking to stay abreast of the latest regulatory considerations and development strategies.
- Virtual Attendance is optimal for an audience who would like to have access to workshop content from anywhere, eliminate travel time and expenses, and who would like to have the flexibility to attend dynamic sessions/presentations at preferred times.
- In-Person Attendance is optimal for an audience that is interested in collaborative discussions and real-time engagement, immersive learning, interactive sessions and group activities.
Registration Fees:
- This workshop is FREE for virtual attendees.
- The combined cost for both days of in-person attendance and activities is:
- $350, in-person attendees – general
- $150, in-person attendees – government (must have an email ending in “.gov” in order to register at this rate)
For faculty and students from the University of Maryland, Baltimore, The Universities at Shady Grove, and University of Michigan, please contact CRCG (info@complexgenerics.org) regarding registration.
Continuing education (CE) credits will not be provided for attending this workshop. A certificate of attendance will only be provided to individuals attending in-person, when requested.
For in-person attendees, beverages and food for breaks will be provided; Lunch will not be provided. Please see the Ordering Lunch tab regarding pre-ordering lunch instructions. Links are provided. Orders must be placed by September 14 (5 PM ET). Also there is an onsite cafe for purchasing food or you are welcome to bring your own lunch.

























 Medical Affairs Manager,
Medical Affairs Manager, 








{kind=link}
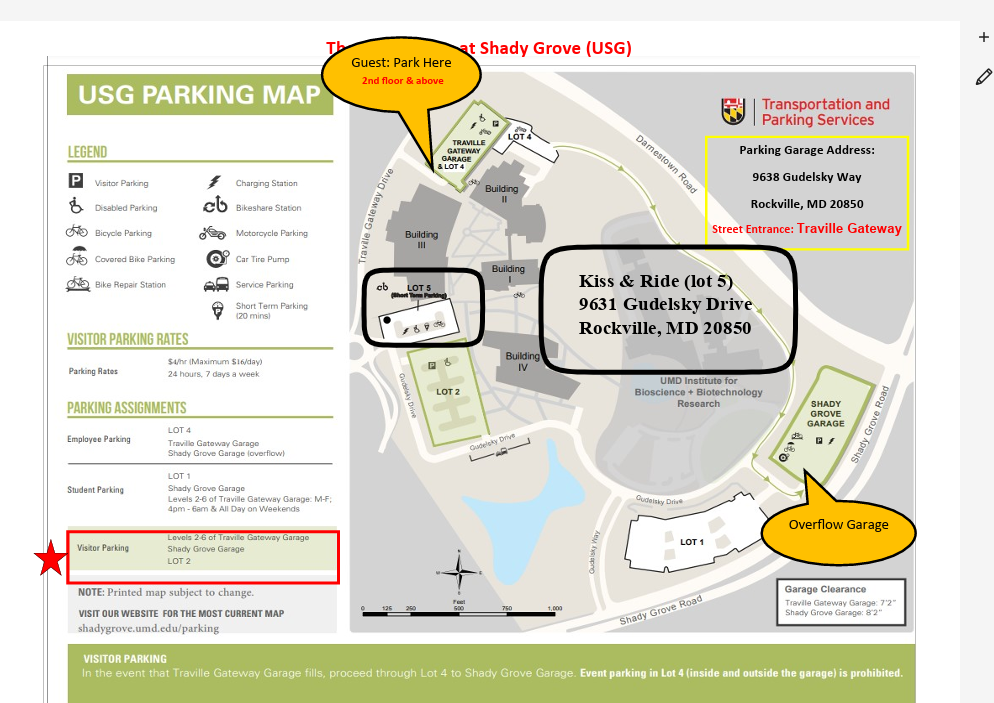{kind=link}